1. p53
在癌症研究领域,p53恐怕是最为人们所熟知的一种抑癌基因。它是细胞生长周期中的一种负调节因子。在癌症应激反应中,P53通过激活多种防御机制防止肿瘤的发生,促进细胞进入细胞周期的停滞阶段,继而凋亡或者衰老。癌细胞中往往出现p53基因突变或者p53蛋白失活。
近日,来自哥伦比亚大学赫伯特·欧文综合癌症中心的一项新的研究发现,当p53蛋白被乙酰化后,它就不能被一种癌蛋白——SET结合并抑制,因此p53活性得以恢复,最终抑制肿瘤生长。这项研究发表于9月14日的Nature杂志上。
- Acetylation-regulated interaction between p53 and SET reveals a widespread regulatory mode. Nature (2016) doi:10.1038/nature19759. Published online 14 September 2016
p53基因是一种抑癌基因,定位于人类染色体17p13.1,编码393个氨基酸组成的53kDa的核内磷酸化蛋白,被称为p53蛋白。p53是一种肿瘤抑制基因(tumor suppressor gene)。在所有恶性肿瘤中,50%以上会出现该基因的突变。由这种基因编码的蛋白质(protein)是一种转录因子(transcriptional factor),其控制着细胞周期的启动。
p53蛋白主要分布于细胞核浆,能与DNA特异结合,其活性受磷酸化、乙酰化、甲基化、泛素化等翻译后修饰调控。正常p53的生物功能好似“基因组卫士(guardian of the genome)”,在G1期检查DNA损伤点,监视基因组的完整性。如有损伤,p53蛋白阻止DNA复制,以提供足够的时间使损伤DNA修复;如果修复失败,p53蛋白则引发细胞凋亡(apoptosis);如果p53基因的两个拷贝都发生了突变,对细胞的增殖失去控制,导致细胞癌变。
2. ApoE
与心血管疾病[2]和阿尔茨海默病[3]有关的载脂蛋白E(ApoE)竟然还是抗癌先锋。这个蛋白不仅可以作用于癌细胞,降低癌细胞的侵袭性;还可以作用于血管内皮细胞,阻止新血管的生成。ApoE基因的表达,就是受肝X受体调控的[7]。肝X受体激动剂GW3965可以促进ApoE基因的表达,抑制动脉粥样硬化的发展[8]。正常细胞合成ApoE蛋白的能力,在癌细胞里面被抑制了。癌细胞表面的LRP1没有见到ApoE就「唆使」癌细胞转移,血管内皮细胞表面的LRP8没有见到ApoE就开启血管生成模式[1]。
证实肝X受体激动剂GW3965可以促进ApoE基因的表达,抑制肿瘤的生长和转移,而且他们还发现,将GW3965与免疫检查点抑制剂CTLA-4抗体等抗癌药物联用,可以大幅提高抗癌效果[9]。
Rgenix理所当然地从葛兰素史克那里获得了GW3965的开发授权,并将其命名为RGX-104[10]。RGX-104除了前面介绍过的功能之外,还可以通过促进ApoE的产生,降低体内免疫抑制细胞“骨髓来源的抑制性细胞(MDSCs)”的水平,解除肿瘤对杀伤性T细胞的免疫抑制。
至此,肝X受体激动剂RGX-104就具备了三大抗癌功能:降低癌细胞的转移性,抑制肿瘤血管的形成,以及解除肿瘤对杀伤性T细胞的免疫抑制。
实际上,MDSCs是癌症等病理因素诱导产生的,它的最主要的特点就是抑制T细胞的能力,促进疾病的发生和进展[13]。而且MDSCs不仅可以直接抑制T细胞的活性,还可以分化成肿瘤相关巨噬细胞和炎性树突细胞[21,22]。
3. MELK
The Maternal Embryonic Leucine Zipper Kinase (MELK) has been identified as a promising therapeutic target in multiple cancer types. MELK over-expression is associated with aggressive disease, and MELK has been implicated in numerous cancer-related processes, including chemotherapy resistance, stem cell renewal, and tumor growth.
MELK expression correlates with tumor mitotic activity but is not required for cancer growth.
More than 30 published articles have suggested that a protein kinase called MELK is an attractive therapeutic target in human cancer, but three recent reports describe compelling evidence that it is not. These reports highlight the caveats associated with some of the research tools that are commonly used to validate candidate therapeutic targets in cancer research.
MELK expression correlates with tumor mitotic activity but is not required for cancer growth. eLife 2018;7:e32838.
Challenges in validating candidate therapeutic targets in cancer. eLife 2018;7:e32402.
4. ALK
不同的ALK融合变异可能会影响患者的生存期。
4.1 什么是ALK融合变异?
我们平常说的ALK阳性,一般是指ALK基因融合,ALK基因一般没有办法单独作恶,它需要和伴侣基因融合才可以作恶。在ALK阳性非小细胞肺癌中,95%均系ALK基因与EML4基因融合。
根据ALK基因与EML4基因融合部位不同,EML4-ALK融合可以分为不同ALK融合变异,其中最常见的是v1变异和v3变异。不同ALK变异比例如下:

值得我们注意的是,免疫组化(IHC)和FISH只能检测ALK融合,不能检测具体的融合变异,PCR和新一代测序(NGS)检测方法才能检测到具体的融合变异。
4.2 v3变异生存期可能缩短
研究人员分析了最常见的v1变异和v3变异对ALK阳性患者生存期的影响,发现v1变异中位生存期(OS)相比v3变异有延长趋势,但无统计学差异,而v1变异患者和v3变异患者的临床特征并无统计学差异。
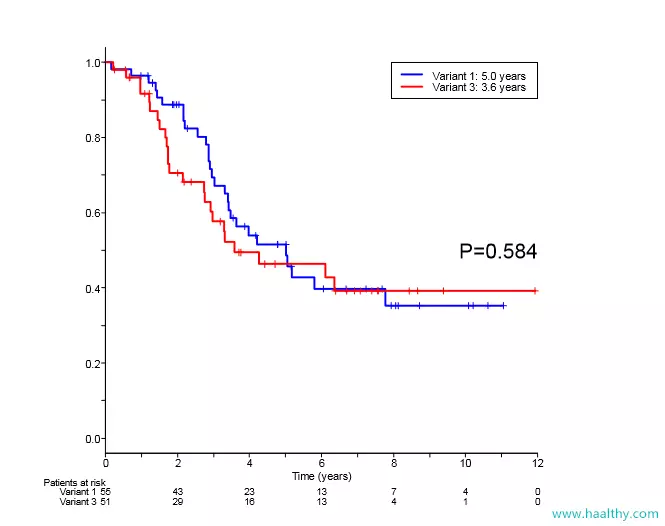
根据上面的研究提示,研究人员又分析了v1变异和v3变异接受一、二、三代ALK靶向药的中位无进展生存期(PFS),发现v1变异接受一、二代ALK靶向药的中位无进展生存期(PFS)相比v3变异都有延长趋势,但未达到统计学差异,唯有接受三代ALK靶向药洛拉替尼时v1变异的中位无进展生存期(PFS)相比v3变异要短。
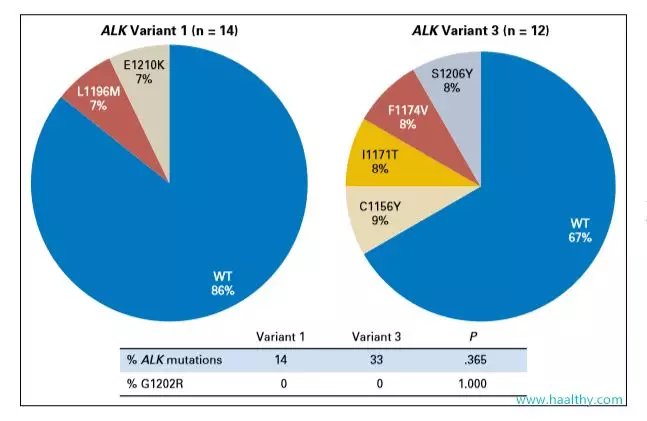
4.3 v3变异更容易出现ALK耐药突变
研究人员发现相比v1变异,v3变异ALK耐药突变发生率更高,尤其是难治的G1202R突变发生率v3变异显著高于v1变异。下图为
克唑替尼耐药后,不同ALK耐药突变发生率:
4.4 洛拉替尼或能延长v3变异生存期
对上述所有回顾性研究发现,v3变异患者更容易发生ALK耐药突变,尤其是难治的G1202R突变,这可能缩短v3变异患者的生存期,而洛拉替尼可以有效抑制G1202R突变,可能更适合ALK阳性患者患者。
- Impact of EML4-ALK Variant on Resistance Mechanisms and Clinical Outcomes in ALK-Positive Lung Cancer. JCO 2018 Jan 26
- http://mp.weixin.qq.com/s?\_\_biz=MzI0MTU4MjEzOA==&mid=2247485122&idx=1&sn=3c1996a618286dd305e3f8bf0949b950&chksm=e9082f97de7fa68162